

("Congenital adrenal hyperplasia," 2010)
The second most common form of CAH is 11-hydroxylase deficiency. A child with this type of CAH has adrenal glands that make too much androgen and not enough cortisol. Children with this type of CAH may also have high blood pressure. These patients do not have aldosterone deficiency.
Rare other types of CAH include 3-betahydroxy-
steroid dehydrogenase deficiency,
lipoid CAH, and 17-hydroxylase deficiency.
Two copies of an abnormal gene are required for disease to occur, and not all mutations and partial deletions result in disease. The phenotype can vary from clinically inapparent disease (occult or cryptic adrenal hyperplasia) to a mild form of disease that is expressed in adolescence or adulthood (nonclassic adrenal hyperplasia) to severe disease that results in adrenal insufficiency in infancy with or without virilization and salt wasting (classic adrenal hyperplasia). The most common form of adrenal hyperplasia (due to a deficiency of 21-hydroxylase activity) is clinically divided into 3 phenotypes: salt wasting, simple virilizing, and nonclassic.
CYP21A is the gene that codes for 21-hydroxylase, CYP11B1 codes for 11-beta-hydroxylase, and CYP17 codes for 17-alpha-hydroxylase. Many of the enzymes involved in cortisol and aldosterone syntheses are cytochrome P450 (CYP) proteins.
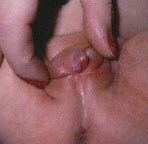
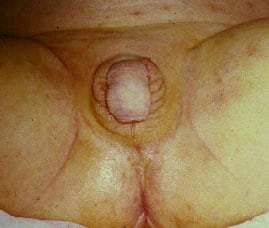
- Clinical presentation in females
- Females with severe forms of adrenal hyperplasia due to deficiencies of 21-hydroxylase, 11-beta-hydroxylase or 3-beta-hydroxysteroid dehydrogenase have ambiguous genitalia at birth due to excess adrenal androgen production in utero. This is often called classic virilizing adrenal hyperplasia.
- Mild forms of 21-hydroxylase deficiency in females are identified later in childhood because of precocious pubic hair, clitoromegaly, or both, often accompanied by accelerated growth and skeletal maturation due to excess postnatal exposure to adrenal androgens. This is called simple virilizing adrenal hyperplasia.
- Milder deficiencies of 21-hydroxylase or 3-beta-hydroxysteroid dehydrogenase activity may present in adolescence or adulthood with oligomenorrhea, hirsutism, and/or infertility. This is termed nonclassic adrenal hyperplasia.
- Females with 17-hydroxylase deficiency appear phenotypically female at birth but do not develop breasts or menstruate in adolescence because of inadequate estradiol production. They may present with hypertension.
- Clinical presentation in males
- 21-hydroxylase deficiency in males is generally not identified in the neonatal period because the genitalia are normal. If the defect is severe and results in salt wasting, these male neonates present at age 1-4 weeks with failure to thrive, recurrent vomiting, dehydration, hypotension, hyponatremia, hyperkalemia, and shock (classic salt-wasting adrenal hyperplasia). Patients with less severe deficiencies of 21-hydroxylase present later in childhood because of the early development of pubic hair, phallic enlargement, or both, accompanied by accelerated linear growth and advancement of skeletal maturation (simple virilizing adrenal hyperplasia).
- In male infants, the disease may be misdiagnosed as gastroenteritis or pyloric stenosis, with potentially disastrous consequences due to delayed treatment with glucocorticoids.
- Males with steroidogenic acute regulatory (StAR) deficiency, classic 3-beta-hydroxysteroid dehydrogenase deficiency, or 17-hydroxylase deficiency generally have ambiguous genitalia or female genitalia because of inadequate testosterone production in the first trimester of fetal life.
- Other findings

- Hyponatremia, hyperkalemia, and/or hypoglycemia suggests the possibility of adrenal insufficiency.
- Hypoglycemia and hypotension may, in part, be due to associated epinephrine synthesis in the adrenal medulla due to cortisol deficiency. Cortisol, perfusing the adrenal medulla from the cortex, normally stimulates phenylethanolamine N -methyltransferase, the last enzyme in epinephrine synthesis.
- Children with simple virilizing 21-hydroxylase deficiency or 11-hydroxylase deficiency have early pubic hair, phallic enlargement, and accelerated linear growth and advanced skeletal maturation.
- Two forms of adrenal hyperplasia (ie, 11-hydroxylase [CYP11B1] and 17-hydroxylase [CYP17] deficiency) result in hypertension due to the accumulation of supraphysiologic concentrations of deoxycorticosterone.2 This weak mineralocorticoid has little consequence at physiologic concentrations but causes sodium retention and hypertension at the supraphysiologic concentrations that occur in these conditions. One form of adrenal hyperplasia results in isolated aldosterone deficiency without affecting the synthesis of cortisol or sex steroids. This form is due to a defect in enzymatic activities that have variously been termed CMO I, CMO II, 18-hydroxylase, or 18-hydroxycorticosterone dehydrogenase; however, it is currently thought to represent one protein called aldosterone synthetase (CYP11B2).
- Other forms of adrenal hyperplasia are characterized by disordered genital development in utero, lack of secondary sexual characteristics development, or hypertension. For example, 17-hydroxylase deficiency in females is rarely identified at birth, but these females seek medical attention later in life because of hypertension or failure to develop secondary sexual characteristics at puberty due to an inability to synthesize estrogens. Male patients with this disorder have ambiguous or female genitalia and may be raised as girls and seek medical attention later in life because of hypertension or a lack of breast development.
- Patients with aldosterone deficiency of any etiology may present with dehydration, hyponatremia, and hyperkalemia, especially with the stress of illness.
- Male or female patients with 11-hydroxylase deficiency may present in the second or third week of life with a salt-losing crisis. However, these patients develop hypertension, hypokalemic alkalosis, or both later in life. This paradox is explained by resistance to mineralocorticoids in infancy and the inability of the elevated deoxycorticosterone levels to replace the deficient serum concentrations of aldosterone in infancy. Upon maturation, mineralocorticoid responsiveness increases, and the elevated concentrations of deoxycorticosterone are sufficient to cause sodium retention, potassium excretion, and hypertension.
- Infants with StAR deficiency (lipoid adrenal hyperplasia) usually have signs of adrenal insufficiency (eg, poor feeding, vomiting, dehydration, hypotension, hyponatremia, hyperkalemia). Some patients do not receive medical attention until late infancy. Male patients with this form of adrenal hyperplasia have female or ambiguous genitalia. Female patients have normal female genitalia. A curious observation is that girls who survive develop breasts and menstruate at puberty, suggesting preservation of ovarian steroidogenesis.
(Wilson, 2009)